
The Responsibilities of the Life Sciences Industry Supplier

Validation is a documented process to prove that equipment, systems, spreadsheets, processes, and procedures work safely and efficiently to protect patients and clients, and to ensure product quality and data integrity.
GAMP5 means Good Automated Manufacturing Practice, is a guide developed by ISPE, and is one of the main references for Life Sciences companies, regarding management of GxP systems.
Most of the automated equipment and systems used by Biopharmaceutical and Medical Device Industries are supplied by third-party companies.
Also, these automated equipment and systems are sold as off-the-shelf systems (i.e., GAMP5 category 3) or configured to meet end-client’s process requirements (i.e., GAMP5 category 4).

Life Sciences companies are highly regulated and are responsible for ensuring that their products are produced with quality and safety.
These companies must maintain the products’ traceability throughout their production cycle while these products are on the market, or throughout the lifetime of a patient (in case of some types of medical products, such as implants).
These companies must generate documentation, known as validation, to comply with regulatory requirements. Moreover, it is necessary to test systems to ensure that they comply with Good Practices and provide the expected results (intended use).
FDA, EMA and ANVISA Requirements
Regulatory agencies such as the FDA, EMA and ANVISA require Life Sciences industries to validate their processes, equipment and systems that impact on product quality and traceability.
The regulated company is responsible for providing documentation during the regulatory agent's audits and inspections. Therefore, it is essential that they have engaged suppliers which meet the requirements of this market.
One may ask, ‘And how does the regulated company ensure that suppliers meet regulatory requirements?’
The answer is: ‘By qualifying suppliers!’
The scope of this qualification may cover architecture, methodology, quality, and validation, to ensure that there is a process consistent enough to meet regulatory requirements and GxP.
The procedure and control applied by the supplier must be appropriate to the level of risk presented by the system.

In this assessment, the following can be checked:
- Implementation and effective running of a Quality and an Information Security Management System;
- Availability of information to support an assessment;
- Understanding of GxP regulations;
- Consistency of the systems development process;
- Frequency and control of updates;
- Cloud usage model (public, hybrid and private);
- Data Integrity and Privacy.
Assessments can be based on available information, questionnaire-based audits, and/or supplier audits (on-line or face-to-face).
Questionnaire-based audits may be appropriate for suppliers of standard products and services. If the supplier is approved, it must be periodically reassessed by the regulated company.
References
Software compliance standards come from a variety of regulatory sources and industry standards that are required in the Life Sciences sector. Serving these standards can be strategic for suppliers' businesses in this market.
The most common references include, but are not limited to:
- Directors' Collegiate Resolution No. 665 from the Brazilian Health Regulatory Agency (ANVISA) which rules on Good Manufacturing Practices of Medical Devices and In-Vitro Diagnostic Products, published on 03/30/2022;
- Directors' Collegiate Resolution No. 430 from the Brazilian Health Regulatory Agency (ANVISA) which rules on Good Distribution, Storage, Transportation Practices of Medicines published on 10/08/2020;
- Directors' Collegiate Resolution No. 48 from the Brazilian Health Regulatory Agency (ANVISA) which rules on Good Manufacturing Practices of personal care products, cosmetics and perfumes published on 10/25/2013.
• ANVISA Guide – Computerized System Validation Guide, No. 33, v. 1, published by ANVISA on 03/26/2020;
• GAMP5 - Good Automated Manufacturing Practice: Computerized System Validation Guide, 2nd edition, published by ISPE in July 2022;
• GAMP® Good Practice Guide: Enabling Innovation, 2021;
• FDA 21 CFR Part 11 - FDA Resolution establishing security policies for the implementation of electronic record and electronic signature for computerized systems to be validated, published in March 1997;
• ISO’s such as: - ISO 13485:2016 (Quality Management Systems); - ISO/IEC 27001:2015 (Information Security Management Systems); - ISO 17025:2017 (Testing and Calibration Laboratories); - ISO 22000:2018 (Food Safety Management Systems); - ISO/TR 80002-2:2017 (Technical Report – Medical Device Software - Part 2: Validation of software for medical device quality systems).
• FDA 21 CFR Part 210 - FDA resolution establishing rules for Good Manufacturing, Processing, Packaging, Retention of Medicines Practices, reviewed in April 2017;
• FDA 21 CFR Part 211 - FDA resolution establishing rules for Good Manufacturing Practices for finished pharmaceutical products, reviewed in April 2017;
• FDA 21 CFR Part 820 - FDA resolution establishing rules to ensure that finished medical devices are safe and effective;
• IEC 62304:2006+AMD1:2015 Defines the life cycle requirements for medical device software;
• NIT-DICLA 038 2019 INMETRO - Application of GLP principles to computerized systems - Brazilian version of OECD No. 17 "Application of GLP Principles to Computerized Systems", 2016;
• OCDE Application of GLP Principles to Computerized Systems - Series in Principles of Good Laboratory Practice and Compliance Monitoring number 17, 2016, for Chemicals, Pesticides and Biotechnology;
• FDA General Principles of Software Validation - FDA (2002), General Principles of Software Validation; Final Guidance for Industry and FDA Staff;
• FDA Guidance for Industry Part 11 - FDA (2003), Guidance for Industry Part 11, Electronic Records; Electronic Signatures — Scope and Application;
• FDA Guidance for Industry Process Validation - FDA (2011), Guidance for Industry - Process Validation: General Principles and Practices;
• FDA 21 CFR Part 106 Infant Formula - FDA (2003), Part 106: Infant Formula Requirements Pertaining to cGMP;
• FDA 21 CFR Part 1271 Human Cells - FDA (2003), Part 106: Infant Formula Requirements Pertaining to cGMP FDA (2004), Part 1271: Human cells, tissues, and cellular and tissue-based products;
• FDA Guide in Food Processing Industry - FDA (2014) Guide to Inspections of Computerized Systems in the Food Processing Industry;
• EudraLex - Volume 4 – Chapter 1: Good Manufacturing Practice (GMP) guidelines, Part Basic Requirements for Medicinal Products, chapter 1 Pharmaceutical Quality System;
• ANNEX 11 EMA - Eudralex – The Rules Governing Medicinal Products in the European Union – Volume 4 – Good Manufacturing Practice – Medicinal Products for Human and Veterinary Use – Annex 11: Computerized Systems;
• ANNEX 15 EMA - EudraLex – The Rules Governing Medicinal Products in the European Union – Volume 4 – Good Manufacturing Practice – Medicinal Products for Human and Veterinary Use – Annex 15: Qualification and Validation;
• GDPR - GDPR, Apr 2016: General Data Protection Regulation;
• Data Integrity Guidelines:
- EMA Questions and Answers – Aug/2016; - FDA Guideline - Data Integrity and Compliance with Drug CGMP – Questions and Answers – Guidance for Industry – Dec/2018; - MHRA Medicine & Healthcare products Regulatory Agency MHRA – GXP Data Integrity Guidance and Definitions – Mar/2018; - PIC/S PI 041-1 Good Practices for Data Management and Integrity in Regulated GMP/GDP Environments – Jul/2021; - WHO Guideline on data integrity, WHO TRS 1033, starting on page 135, 2021 • WHO Technical Report Series 1033 - WHO Expert Committee on Specifications for Pharmaceutical Preparations, 2021, Fifty-fifth report;
• WHO Guidelines on Validation - Guidelines on Validation – Appendix 5 Validation of computerized Systems (August 2018) – Draft for Comments;
• IMDRF International Medical Device Regulators Forum: - UDI guidance, Dec/2013; - UDI Application Guide, Mar/2019; - Audit report, Oct/2015; - SaMD Clinical Evaluation, Sep/2015; - Essential Principles of Safety and Performance of Medical Devices and IVD (Vitro Diagnostic Product), Oct/2018; - Principles and Practices for Medical Device Cybersecurity, Mar/2020. • ICH Good Clinical Practices - ICH E6 (R2) Good Clinical Practices – Nov/2016;
• ICH Good Manufacturing Practices - ICH Q7 Good Manufacturing Practices for Active Ingredients – Nov/2000;
• PIC/S Good Practices for Computerized Systems - PIC/S Guidance PI011-3, Sep/2007 Good Practices for Computerized Systems in Regulated “GXP” Environments.
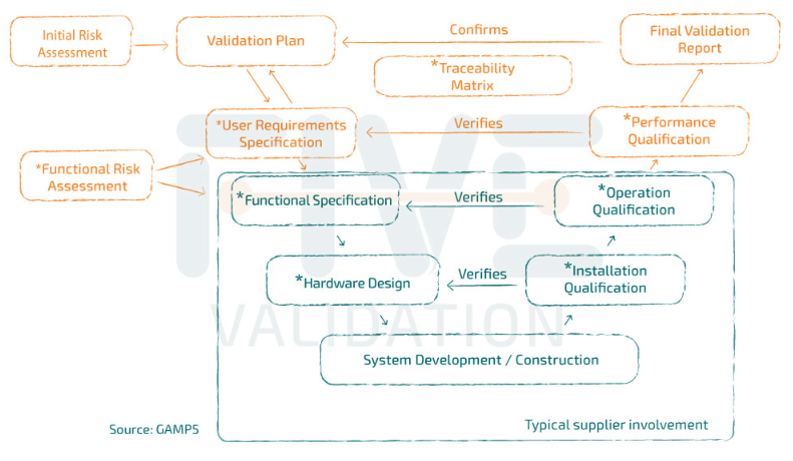
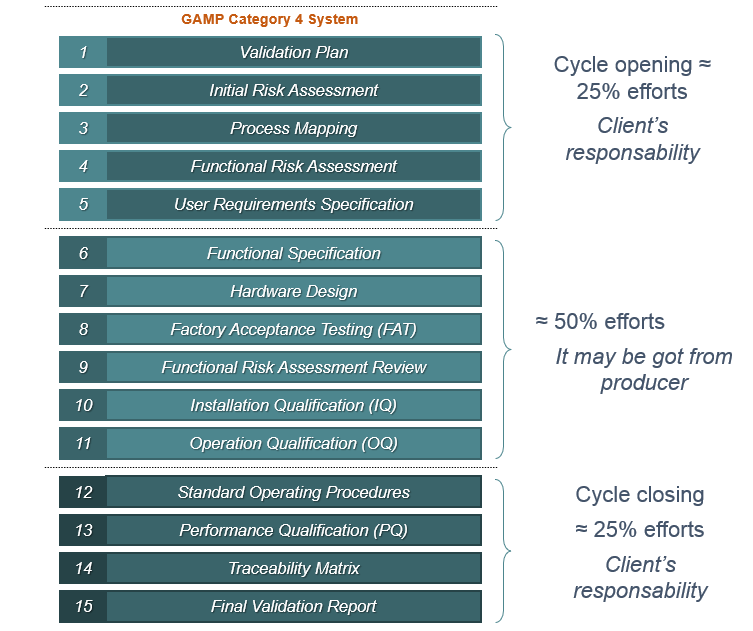
Typical Supplier Documentation
Even when an industry chooses to purchase packages of validation or qualification documents provided by the equipment or system manufacturers themselves, it is important to remember that these packages are partial.
This means that it is the industry's responsibility to review what has been delivered to them and still develop the remaining part of the cycle to complete the validation or qualification study, as recommended by the regulatory bodies.
When Does Validation Start?
Computerized systems in the cloud need to meet the same regulatory rules as systems outside the cloud if they are GxP relevant.
The validation team should be involved BEFORE making the purchase, so it is important that there is documentation at the supplier qualification stage.
Cloud Supplier’s Responsibilities
- Infrastructure needed to make services available (servers, connectivity, and information security);
- Data retention for the minimum period in accordance with the regulated company's GMP, including backup and restoration;
- Application of best information security practices to prevent cyber-attacks and data leaks;
- The SaaS partner becomes as critical as the raw material supplier because it is now these companies that retain the industry's data (traceability and data integrity).
- Comply with the same best practices expected to serve the client, as far as they apply to the supplier's business.
Even if the supplier is not subject to FDA, EMA or ANVISA regulation, it is essential that they adopt the same practices as the industry in managing and maintaining the system and data under their responsibility.
This initiative, although not a regulatory requirement that directly affects the supplier, is fundamental from a commercial point of view and can be decisive in the choice of a supplier by companies in the sector.
The scarcity of suppliers who adopt such practices can be a differentiator when prospecting for new clients, especially for Life Sciences companies, whose services have high added value and a high average ticket.
Vendors which consolidate in this market may have a significant financial return.
SaaS Suppliers of GxP Applications
It is strategic and relevant for a SaaS provider of relevant GxP application to validate the standard functions of its software before deploying it to a production environment.
With each new function or improvement, it is necessary to validate this release to identify possible bugs, deviations, and impacts, as well as providing clear and transparent release notes on the changes made.
Where to start?
To achieve a degree of consistency and maturity in the development and quality system, we can provide training and consultancy, as well as our validation services.
FIVE also presents itself as a partner to the industry and supplier, making itself available to assist them in reviewing their quality system, cloud infrastructure qualification and documentation, as well as completing the validation or qualification cycle.
It is essential to adopt these good practices as a "lifestyle", as they will recur. With each new version or change, documentation needs to be updated.
And with this continuous process in mind, it's practically impossible to do an efficient job without a digital validation tool.
Click here to find out about GO!FIVE®, cloud validation software that is adapted to apply the Agile Framework, in compliance with the FDA, EMA, WHO and ANVISA, making it an excellent option for phased implementations.
Click here to read our article on how to apply critical and agile thinking and IT service management in relation to the life sciences sector.
Provider of solutions on premise or in the clients’ cloud
In this article, we covered the responsibilities of suppliers, especially those offering cloud systems. However, many of the ideas discussed in this text are also relevant to cases in which the client is responsible for the infrastructure.
It is in the system acquisition phase that the documents that will be the supplier's scope are usually included.
It is the supplier's role to provide documents that facilitate and speed up the validation process. However, it is important to note that the responsibility for "opening" and "closing" the validation cycle lies with those who will be using the system, in conjunction with Quality Assurance.
References:
-
- GAMP5 - Good Automated Manufacturing Practice: Computerized System Validation Guide, second edition, published by ISPE in July 2022.
- ANVISA Guide - Computerized System Validation Guide, nº 33, version 1, published by ANVISA, 26/03/2020.
- https://usdm.com/resources/blogs/how-do-life-science-companies-qualify-vendors-and-software.



{kind=link}
{kind=link}
{kind=link}