
Responsabilidades de los proveedores del sector de las Ciencias de la Vida

La validación es un proceso documentado para demostrar que los equipos, sistemas, hojas de cálculo, procesos y procedimientos funcionan con seguridad y eficacia para proteger a pacientes y consumidores, y garantizar la calidad del producto y la integridad de los datos.
GAMP5 es una guía de Buenas Prácticas de Fabricación (BPF) Automatizada, elaborada por ISPE, que constituye una de las referencias clave para las empresas de Ciencias de la Vida en la gestión de sus sistemas BPx relevantes (impacto en el producto).
La mayoría de los equipos y sistemas automatizados utilizados por las industrias biofarmacéuticas y de productos sanitarios son suministrados por empresas terceras y vendidos como sistemas listos para usar (categoría 3 de GAMP5), o configurados para cumplir los requisitos de proceso del cliente final (categoría 4 de GAMP5).

Las empresas de Ciencias de la Vida están sometidas a un alto grado de regulación y son responsables de garantizar que sus productos se fabrican con calidad y seguridad y que se mantiene la trazabilidad durante todo su ciclo de producción, mientras están en el mercado o, en el caso de algunos tipos de productos médicos como los implantes, durante toda la vida del paciente.
Para cumplir los requisitos normativos, estas empresas deben generar documentación, conocida como validación, y someter a prueba sus sistemas para garantizar que se ajustan a las buenas prácticas y producen los resultados esperados (uso previsto).
Requisitos de FDA, EMA y ANVISA
Los organismos reguladores, como FDA, EMA y ANVISA, exigen a estas industrias que validen sus procesos, equipos y sistemas que afectan la calidad y trazabilidad de los productos. La empresa regulada es responsable de proporcionar esta documentación durante las auditorías e inspecciones del agente regulador.
Por tanto, es esencial contar con proveedores comprometidos que comprendan los requisitos de este mercado.
¿Y cómo se asegura la empresa regulada de que los proveedores cumplen los requisitos normativos?
Mediante la calificación de los proveedores.
El alcance de esta calificación puede abarcar la arquitectura, la metodología, la calidad y la validación para garantizar que existe un proceso lo suficientemente sólido como para cumplir los requisitos reglamentarios y que sigue las mejores prácticas.
Este procedimiento y el control aplicado por el proveedor deben ser adecuados al nivel de riesgo presentado por el sistema.

En esta evaluación se puede comprobar lo siguiente:
- Implementación y buen funcionamiento del sistema de gestión de calidad y seguridad de la información;
- Disponibilidad de información para apoyar una evaluación;
- Conocimiento de la normativa BPx;
- Solidez del proceso de desarrollo de sistemas;
- Frecuencia y control de las actualizaciones;
- Modelo de uso de la nube (pública, híbrida y privada);
- Integridad y privacidad de los datos.
Las evaluaciones pueden basarse en la información disponible, en auditorías basadas en cuestionarios y en auditorías de proveedores (en línea o presenciales).
Las auditorías basadas en cuestionarios pueden ser adecuadas para los proveedores de productos y servicios estándares.
Si el proveedor es aprobado, debe ser reevaluado periódicamente por la empresa regulada.
Fuentes de referencia
Las normas de conformidad del software proceden de diversas fuentes normativas y estándares industriales que se exigen en el sector de las Ciencias de la Vida y cuyo cumplimiento puede ser estratégico para el negocio de los proveedores de este nicho de mercado.
Las fuentes más comunes incluyen, entre otras:
- Resolución del Consejo Colegiado nº 665 de la Agencia Nacional de Vigilancia Sanitaria de Brasil que aprueba el reglamento técnico de Buenas Prácticas de Fabricación de Productos Médicos y Productos para Diagnóstico de Uso in Vitro, publicada el 30/03/2022;
- Resolución del Consejo Colegiado nº 430 de la Agencia Nacional de Vigilancia Sanitaria de Brasil que dispone sobre las Buenas Prácticas de Distribución, Almacenamiento y Transporte de Medicamentos, publicada el 08/10/2020;
- Resolución del Consejo Colegiado nº 48 de la Agencia Nacional de Vigilancia Sanitaria de Brasil que determina las Buenas Prácticas de Fabricación de productos de higiene personal, cosméticos y perfumes publicada el 25/10/2013.
• Guía de ANVISA - Guía para Validación de Sistemas Informáticos, nº 33, versión 1, publicado por ANVISA en 26/03/2020.
• GAMP5 - Good Automated Manufacturing Practice: Guía de Validación de Sistemas Informáticos, segunda edición, publicado por ISPE en julio de 2022.
• GAMP™ Good Practice Guide: Enabling Innovation, 2021.
• FDA 21 CFR Part 11 - Resolución de FDA por la que se establecen las políticas de seguridad para la implantación del registro electrónico y la firma electrónica para los sistemas informáticos que deben validarse, publicada en marzo de 1997.
• ISOs como, por ejemplo:
- ISO 13485:2016 (Gestión de Sistemas de Calidad para Fabricantes de Productos Sanitarios);
- ISO/IEC 27001:2015 (Gestión de Seguridad de la Información);
- ISO 17025:2017 (International Standard : General requirements for the competence of testing and calibration laboratories);
- ISO 22000:2018 (International Standard – Food Safety Management Systems);
- ISO/TR 80002-2:2017 (Technical Report : Medical Device Software - Part 2: Validation of software for medical device quality systems).
• FDA 21 CFR Part 210 - Resolución de FDA por la que se establecen reglas para Buenas Prácticas de Fabricación, tratamiento, envasado y conservación de los medicamentos, revisada en abril de 2017.
• FDA 21 CFR Part 211 - Resolución de FDA por la que se establecen normas de buenas prácticas de fabricación de productos farmacéuticos acabados, revisada en abril de 2017.
• FDA 21 CFR Part 820 - Resolución de FDA que establece normas para garantizar que los productos médicos acabados sean seguros y eficaces.
• IEC 62304:2006+AMD1:2015 Define el ciclo de vida de los requisitos para software de productos sanitarios.
• NIT-DICLA 038 2019 INMETRO - Aplicación de los principios BPL a los sistemas informáticos - versión brasileña de OCDE n.º 17 “Application of GLP Principles to Computerized Systems”, 2016.
• OCDE Application of GLP Principles to Computerized Systems - Series in Principles of Good Laboratory Practice and Compliance Monitoring number 17, 2016, for Chemicals, Pesticides and Biotechnology.
• FDA General Principles of Software Validation - FDA (2002), General Principles of Software Validation; Final Guidance for Industry and FDA Staff.
• FDA Guidance For Industry Part 11 - FDA (2003), Guidance for Industry Part 11, Electronic Records; Electronic Signatures — Scope and Application.
• FDA Guidance For Industry Process Validation - FDA (2011), Guidance for Industry - Process Validation: General Principles and Practices.
• FDA 21 CFR Part 106 Infant Formula - FDA (2003), Part 106: Infant Formula Requirements Pertaining to cGMP.
• FDA 21 CFR Part 1271 Human Cells - FDA (2003), Part 106: Infant Formula Requirements Pertaining to cGMP FDA (2004), Part 1271: Human cells, tissues, and cellular and tissue-based products.
• FDA Guide in Food Processing Industry - FDA (2014) Guide to Inspections of Computerized Systems in the Food Processing Industry.
• EudraLex - Volume 4 – Chapter 1: Good Manufacturing Practice (GMP) guidelines, Part Basic Requirements for Medicinal Products, chapter 1 Pharmaceutical Quality System.
• ANNEX 11 EMA - Eudralex – The Rules Governing Medicinal Products in the European Union – Volume 4 – Good Manufacturing Practice – Medicinal Products for Human and Veterinary Use – Annex 11: Computerized Systems.
• ANNEX 15 EMA - EudraLex – The Rules Governing Medicinal Products in the European Union – Volume 4 – Good Manufacturing Practice – Medicinal Products for Human and Veterinary Use – Annex 15: Qualification and Validation.
• GDPR - GDPR, Apr 2016: General Data Protection Regulation.
• Directrices de Integridad de Datos.
- EMA Questions and Answers – Ago/2016;
- FDA Guideline - Data Integrity and Compliance with Drug CGMP – Questions and Answers – Guidance for Industry – Dez/2018;
- MHRA Medicine & Healthcare products Regulatory Agency MHRA – GXP Data Integrity Guidance and Definitions – Mar/2018;
- PIC/S PI 041-1 Good Practices for Data Management and Integrity in Regulated GMP/GDP Environments – Jul/2021;
- WHO Guideline on data integrity, WHO TRS 1033, starting on page 135, 2021.
• WHO Technical Report Series 1033 - WHO Expert Committee on Specifications for Pharmaceutical Preparations, 2021, Fifty-fifth report.
• WHO Guidelines on Validation - Guidelines on Validation – Appendix 5 Validation of computerized Systems (August 2018) – Draft For Comments.
• IMDRF International Medical Device Regulators Forum.
- UDI guidance, Dec/2013;
- UDI Application Guide, Mar/2019 ;
- Audit report, Oct/2015 ;
- SaMD Clinical Evaluation, Sep/2015;
- Essential Principles of Safety and Performance of Medical Devices and IVD (Vitro Diagnostic Product), Oct/2018;
- Principles and Practices for Medical Device Cybersecurity, Mar/2020.
• ICH Good Clinical Practices - ICH E6 (R2) Good Clinical Practices – Nov/2016.
• ICH Good Manufacturing Practices - ICH Q7 Good Manufacturing Practices for Active Ingredients – Nov/2000.
• PIC/S Good Practices for Computerized Systems - PIC/S Guidance PI011-3, Set/2007 Good Practices for Computerized Systems in Regulated “GXP” Environments.
Documentación típica preparada por el proveedor
Incluso cuando una industria opta por adquirir paquetes de documentos de validación o calificación puestos a su disposición por los propios fabricantes de equipos o sistemas, es importante recordar que estos paquetes son parciales, o sea, es responsabilidad de la industria revisar lo entregado y seguir desarrollando parte del ciclo que queda para completar el estudio de validación o calificación, tal y como recomiendan los organismos reguladores.
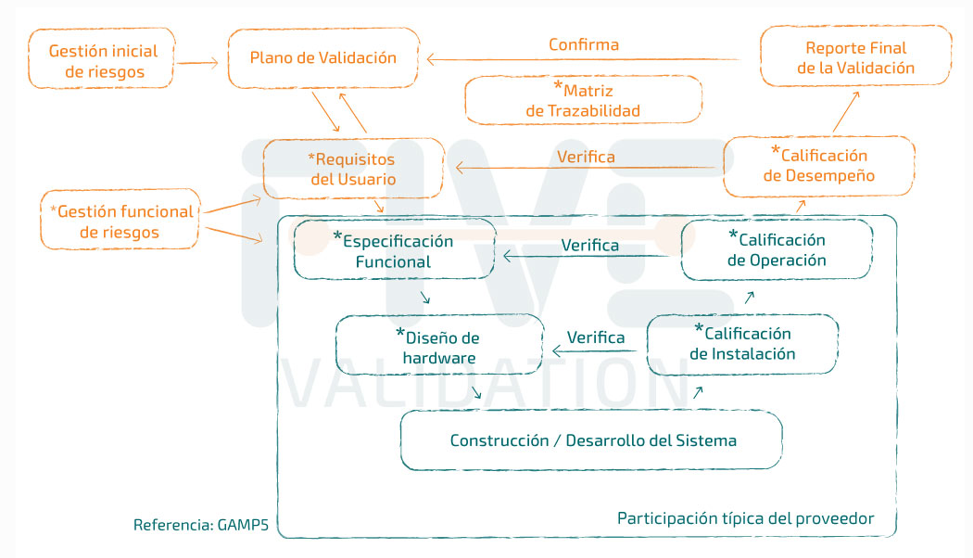
¿Cuándo empieza la validación?
El sistema informático en la nube debe cumplir las mismas normas reglamentarias que los sistemas fuera de la nube, si fueran BPx relevantes.
El equipo de validación debe participar ANTES de la adquisición, por lo que la documentación en la fase de calificación del proveedor es importante.
Responsabilidades del proveedor de la nube
- Infraestructura necesaria para prestar los servicios (servidores, conectividad y seguridad de la información);
- Conservación de los datos durante el periodo mínimo de acuerdo con las BPF de la empresa regulada, incluyendo copias de seguridad y restauración;
- Aplicación de las mejores prácticas de seguridad de la información para evitar ciberataques y fugas de datos;
- El proveedor SaaS pasa a ser tan crítico como el de materias primas, porque ahora, son las empresas que empiezan a retener los datos de la industria (trazabilidad e integridad de los datos);
- Cumplir con las mismas buenas prácticas previstas para el cliente, en lo que es aplicable al negocio del proveedor.
Aunque el proveedor no esté sujeto a la reglamentación de FDA, EMA y ANVISA, es esencial que adopte las mismas prácticas del sector para la gestión y el mantenimiento del sistema y los datos bajo su responsabilidad.
Esta iniciativa, aunque no es un requisito reglamentario que afecte directamente al proveedor, es fundamental desde el punto de vista comercial y puede ser decisiva en la elección de un proveedor por parte de las empresas del sector.
La escasez de proveedores que adopten estas prácticas puede ser un diferencial en la prospección de nuevos clientes, especialmente para las empresas de Ciencias de la Vida, cuyos servicios tienen un alto valor añadido y un tique medio elevado.
Consolidarse en este mercado puede generar un importante retorno financiero para los proveedores.
.

Proveedores de SaaS de aplicaciones BPx
Es estratégico y relevante para un proveedor de SaaS de aplicaciones BPx validar las características estándares de su software antes de implementarlo (deploy) en un entorno de producción.
Con cada nueva función o mejora, es necesario validar esta versión para identificar posibles errores, desviaciones e impactos, además de proporcionar notas de versión claras y transparentes sobre los cambios realizados.
¿Por dónde empezar?
Para alcanzar un grado robusto y maduro de sistema de calidad y desarrollo, podemos proporcionar formación y consultoría, así como nuestros servicios de validación.
FIVE también se presenta como parcero de la industria y de los proveedores, poniéndose a su disposición para ayudarles a revisar su sistema de calidad, la calificación y documentación de la infraestructura en la nube, así como a completar el ciclo de validación o calificación.
Es fundamental adoptar estas mejores prácticas como un "estilo de vida", ya que serán recurrentes. Con cada nueva versión o cambio, es necesario actualizar la documentación.
Y teniendo en cuenta esta continua rutina, es casi imposible realizar un trabajo eficiente sin una herramienta de validación digital.
PINCHE AQUÍ y conozca GO!FIVE®, un software de validación en la nube que está adaptado para aplicar el framework Agile, cumple con la FDA, la EMA, la OMS y ANVISA, y es una excelente opción para implementaciones escalonadas.
PINCHE AQUÍ y consulte nuestro artículo sobre cómo utilizar el pensamiento crítico, la agilidad y la gestión de servicios de TI en relación con el sector de las Ciencias de la Vida..
Proveedor de soluciones on premise o en la nube del cliente
En este artículo hemos abordado las responsabilidades de los proveedores, en particular de los que ofrecen sistemas en la nube. No obstante, muchas de las ideas tratadas en este texto también son pertinentes para los casos en los que el cliente es responsable de la infraestructura.
Es en la fase de adquisición del sistema cuando suelen incluirse los documentos que serán del alcance del proveedor.
Es función del proveedor proporcionar documentos que faciliten y agilicen el proceso de validación. Sin embargo, es importante subrayar que la responsabilidad de "abrir" y "cerrar" el ciclo de validación recae en quienes utilizarán el sistema, junto con el Control de Calidad.
Referencias:
- GAMP5 - Good Automated Manufacturing Practice: Guía de Validación de Sistemas Informáticos, segunda edición, publicado por ISPE en julio de 2022.
- Guía de ANVISA - Guía de Validación de Sistemas Informáticos, nº 33, versión 1, publicado por ANVISA, 26/03/2020
- https://usdm.com/resources/blogs/how-do-life-science-companies-qualify-vendors-and-software



{kind=link}
{kind=link}
{kind=link}