
What is Computer System Validation – CSV?

From day one,
agile compliance
that empowers
your team!
What is Computer System Validation - CSV?
The validation proves with documentation that computer systems used in industrial production adequately fulfill their automatic functions and contribute to ensure the traceability of produced batches and meets GMP regulations. Every computer system that has a direct or indirect relation to the production of the medicine, product for health or impact on traceability, must be validated, because it is a GxP system.
The first important activity of the Computer Systems Validation professional is to gather all the available documentation about the system which is object of the study.
Validation based on risk approach
The study of the existing documentation to survey the risk scenarios is extremely important to know the particularities of the system to be validated and to meet cGMP guidelines. However, after some systems have been validated by the professional, some risk scenarios are invariably presented, such as the system behavior facing power outage, study of access profiles, access security, quality of electronic record produced by the system including audit trail, application of electronic signatures, etc.

It is important to emphasize that Functional Risk Assessment must be done by a multidisciplinary team due to the need of adopting mitigation measures for the risks which result in "medium" and "high" levels. The acceptance of these mitigation measures must be agreed among the team aiming to really be functional, whether they are adopting new work procedures or improvements in the system. The measures to be adopted become the validation strategy and they are the main points where GxP validation should focus.
* For the Functional Risk Assessment, the participation of the system supplier or developer is recommended. When this is not available, a professional can be hired with knowledge in this type of system that is being assessed.
Why do I need to validate?
In addition to contributing for data quality and data integrity, the GMP validation lifecycle is valuable because it allows:
• Extracting all the needed available resources in the system to cover the specific process safely;
• Make the technical knowledge team more in-depth about the system, avoiding that the knowledge stays entirely on the hands of the supplier (opening the "black box");
• Document technical discoveries, avoiding the loss of knowledge in an eventual exit of professionals of the company (risk to the business!);
• Explore all possible automatic features in order to avoid manual steps, leading to repeatability and reproducibility for the process (target of validation!);
• Direct the team to analyze necessary actions and documented procedures for contingency planning, data backup and application, and disaster recovery, reducing production downtime (risk to the business!);
• Reliability of process information avoiding operational errors.
Learn more about Validation Life Cycle
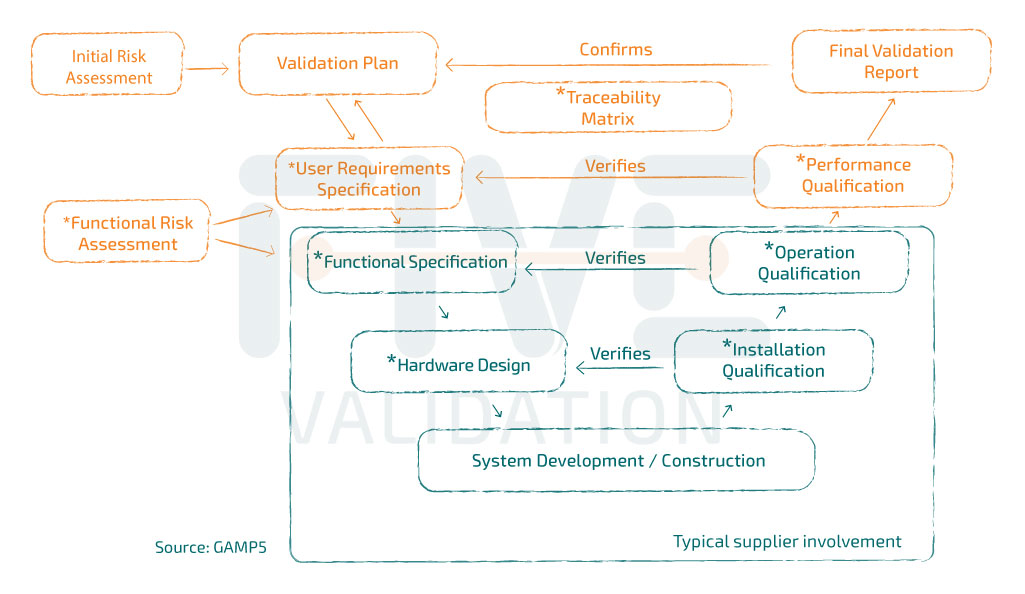
GAMP5® Validation Guide
GAMP5® is a Good Automated Manufacturing Practice guide, the 5th version of 1st edition was released in 2008. Recently, in 2022, they published the 2nd edition of the guide. Since then, it has revolutionized the Validating Computer Systems method.
The guide is the main source of "inspiration" for Computer Validation professionals and has as its central axis the risk-based validation strategy ("A Risk-Based Approach to Compliant GxP Computerized Systems").
Note.: although each company has its specificity, there are several standard processes within a type of system, for example purchasing processes within an ERP, which are common to different companies. Taking this opportunity, FIVE has developed a paperless e-validation software named GO!FIVE®, where users can select multiple libraries, according to the process or system they want to validate, and these libraries contain good market practices, regulatory requirements and much more. Click here and learn more about Paperless Validation Software.
GxP is a general term for the application of good practices. The 'x' indicates the area in which good practices are related (manufacturing, distribution, clinical research, laboratory, etc.). The relevant GxP system is any and all system that has impact on:
- Patient health;
- Product quality;
- Data integrity.
The best way to validate is undoubtedly, risk-based, whether it is a new system (prospective validation) or a legacy system (concurrent validation). If the risk results in 'medium' and 'high' levels, a mitigation measure should be envisaged. If mitigation or upgrade isn’t possible, system exchange should be considered.

URS – User Requirement Specification
Normally, these "medium" and "high" risk mitigation measures are detailed in the User Requirement Specification (URS), which becomes the reference document for system validation. If it is a non-configurable system, tests should basically be drawn up to prove that URS requirements have been met and some other typical system tests covered in the following GMP testing phases:
- Installation Qualification;
- Operational Qualification;
- Performance Qualification.
If the system is configured or customized to meet the needs of the user company, specification documents must be produced. Some examples:
- Functional Specification;
- Hardware Design;
- Software Design.
Classification / Categorization of Computerized Systems
| GAMP® | Comentário |
|---|---|
| Category 1 | Infrastructure Software |
| Category 3 | Non configurable product |
| Category 4 | Configured product |
| Category 5 | Custom application |
• It is important to note that acquiring the vendor's document package doesn’t reflect the completion of the validation work. It is necessary to "open" the document life cycle, with the issuance of the Validation Plan, Functional Risk Assessment and URS and "close" the lifecycle with the issuance of the performance test protocol, Traceability Matrix and Validation Report, documents that are not normally part of the solution provider's scope.

FDA 21 CFR Part 11
It is the FDA standard that establishes rules for use of electronic registration in the Life Sciences industries. In a very brief way, the system must broaden:
• Electronic tamper-proof files or database to ensure data integrity
• Audit Trail
• Access control
• Electronic signature = ID + password
• Accounts use guarantee by its genuine users
• A ppropriate methods to prevent unauthorized access.
The requirements of FDA 21 CFR Part11 are currently quite common in the market and if foreseen at the beginning of the project, it brings safety and necessary traceability for the good use of the system. For legacy systems that do not comply with the regulation, the best way is the Risk Analysis. We suggest studying precisely the absence of these electronic security.

Data Integrity
One of the key requirements for the computer system is to ensure that the generated data in production is complete from the beginning to the end of the process.
It means that the system must be able to keep records of who entered the system, when, what was the action, why it was done and where it was done.
Normally, the impact on data integrity is primarily related to this production batch data traceability (depending on the validation focus system).
Click here for more information on Data Integrity in the Pharmaceutical Industry.
Click here and know our Paperless Validation Software
GAMP5® is a guide that has its intellectual rights reserved by ISPE™. Available for purchase at ispe.org



{kind=link}
{kind=link}
{kind=link}