
O que é UDI (Identificação Única de Dispositivos)? Princípios para projeto e operação de sistemas – dispositivos médicos

O que é UDI (Identificação Única de Dispositivos)?
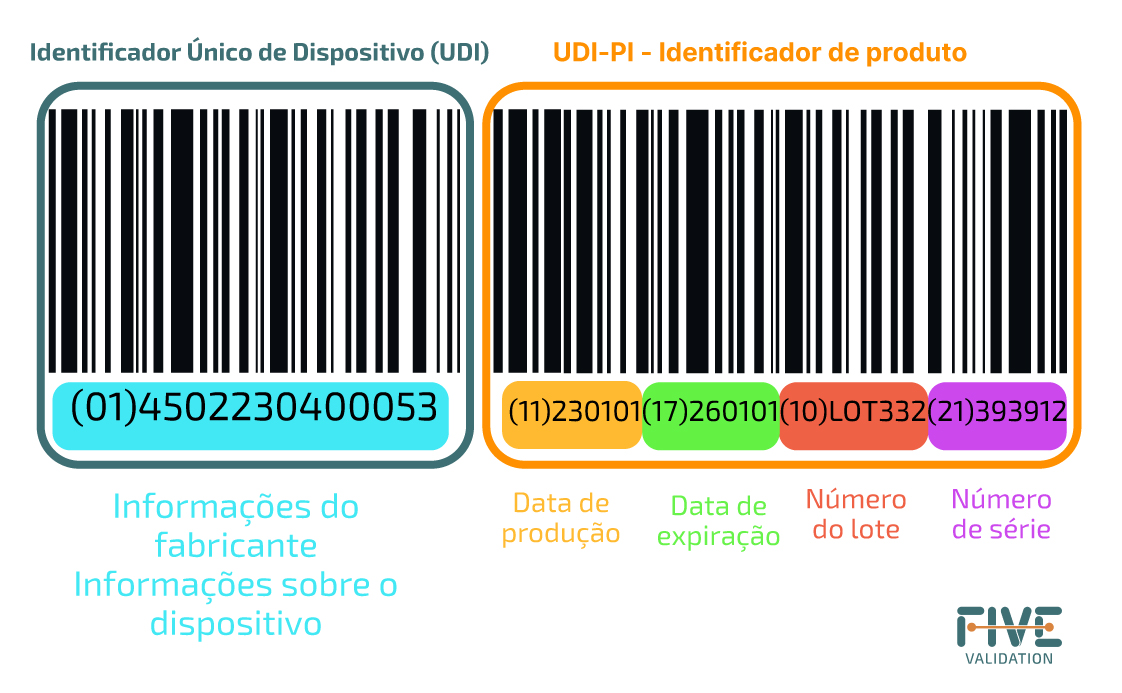
Obs.: PI (Identificador de Produção) é um código variável que representa um ou mais dos seguintes itens: número de série do dispositivo; número de lote; data de validade do dispositivo; data de fabricação do dispositivo apresentada em formato padrão (AAAA-MM-DD); Código de identificação no caso de produto baseado em células humanas (HCT/P).
A figura abaixo descreve as etapas da implementação da UDI.

Os padrões e prazos da UDI variam de acordo com o mercado e o produto, e foram revisados diversas vezes em alguns países.
Embora na Europa as diretrizes tenham começado com a publicação do IMDRF em 2013, que discutiremos mais adiante, isto não era novidade para a indústria de dispositivos médicos, uma vez que tem sido uma necessidade para produtos exportados para os EUA há muitos anos.
Na Europa, o número UDI também será um requisito para o rastreamento de produtos, para o qual deverá ser utilizada a base de dados EUDAMED.
Ao contrário dos EUA e do Brasil, é necessário um Basic UDI-DI adicional para a Europa. O Basic-UDI-DI (Basic Unique Device Identification - Device Identifier) é um identificador de agrupamento para dispositivos médicos com o mesmo uso pretendido, classe de risco e características essenciais de projeto e fabricação. Ao contrário do UDI-DI, que é específico para cada modelo e versão do dispositivo, o Basic-UDI-DI não aparece no rótulo ou na embalagem do produto. Em vez disso, serve como um identificador chave em bases de dados e documentação regulamentares, garantindo a rastreabilidade e facilitando os processos regulamentares.
Importante: a empresa regulada deve garantir que os sistemas UDI cumprem os regulamentos apropriados. Este é o papel do processo de validação. Explore nossa página de serviços e descubra como a FIVE Validation pode aprimorar seu trabalho!
International Medical Device Regulators Forum (IMDRF)
O que está no rótulo?
Alguns dos números entre colchetes no componente UDI-PI serão reconhecíveis. Cada um desses números transmite detalhes específicos sobre o item:
(01) GTIN
(10) é para o número do lote.
(11) é para a data de produção.
(17) define a data de vencimento.
(21) é o número de série.
Isso significa que a cada novo lote ou data de validade atualizada todos esses detalhes serão alterados, tornando o código dinâmico.
O UDI é composto por duas partes:
Identificador de Dispositivo (UDI-DI)
+
Identificador de Produção (UDI-PI)
a) O número do lote dentro do qual um dispositivo foi fabricado;
b) O número de Série de um dispositivo específico;
c) A data de validade de um dispositivo específico;
d) A data de fabricação (pode não ser exigida caso existam outros Identificadores de Produção no rótulo);
e) A versão, para Software como Dispositivo Médico (SaMD),
Obs.: A versão SaMD poderá ser capturada no Identificador de Produção do lote sob determinadas regulamentações nacionais.
f) O Código de Identificação Distinta (DIC), quando aplicável. Este número é um identificador essencial para produtos médicos de origem humana
Obs.: Os produtos médicos de origem humana (MPHO) incluem sangue, órgãos, medula óssea, sangue do cordão umbilical, córneas, tecidos, células reprodutivas e leite derivado de humanos para uso terapêutico. O uso de DIC em UDI é limitado a produtos MPHO regulamentados como dispositivos médicos.
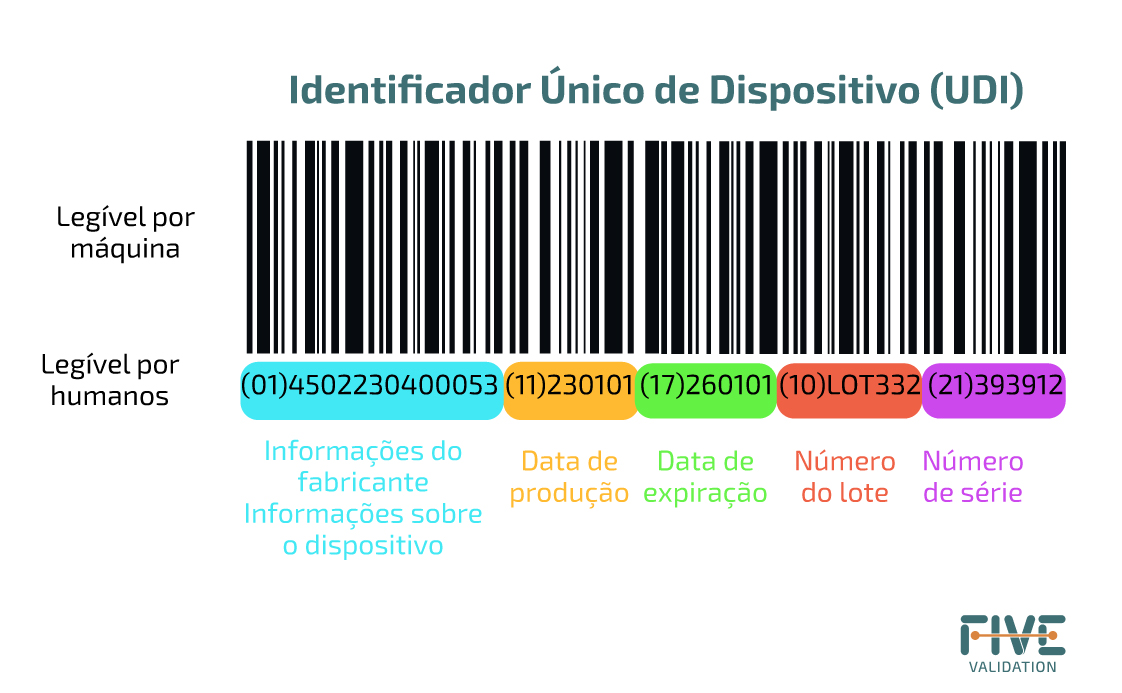
a) Identificação Automática e Captura de Dados (AIDC) – é uma tecnologia utilizada para captura automática de dados. As tecnologias AIDC incluem códigos de barras, cartões inteligentes, biometria e RFID (identificação por radiofrequência).
b) Interpretação Legível por Humanos (HRI) – é uma interpretação legível dos caracteres de dados codificados no suporte UDI. É importante caso não exista um sistema automático de leitura do código.
O suporte UDI, portanto, deve inicialmente ter uma seção que possa ser lida automaticamente, como um código de barras, e ser seguida por uma parte que possa ser lida por humanos, como códigos numéricos.
Quando o sistema UDI estiver totalmente implementado, a etiqueta da maioria dos dispositivos incluirá uma UDI em formato legível e um suporte AIDC.
O sistema UDI destina-se a ser um identificator também utilizado no contexto de transações comerciais e clínicas, incluindo a rastreabilidade de dispositivos no ambiente pós-comercialização (por exemplo, ordens de compra, faturas, manutenção/gestão de inventário, notas clínicas, etc.).
O suporte da UDI deve ser colocado no rótulo do dispositivo e em todos os níveis superiores de embalagem e, no caso de dispositivos reutilizáveis, no próprio dispositivo (marcação direta).
Gostaria de saber mais sobre a função de validação no projeto UDI? Por favor, entre em contato com um de nossos especialistas em [email protected]
Software as a Medical Device, SaMD - critérios de atribuição UDI
A UDI deve ser atribuída ao nível do sistema do Software as Medical Device (SaMD).
O número da versão do SaMD é considerado o mecanismo de controle de fabricação e deve ser exibido no UDI-PI.
A seguinte alteração de um SaMD exigiria um novo UDI-DI:
- As principais revisões do SaMD serão identificadas com um novo UDI-DI;
- As principais revisões do SaMD são entendidas como alterações complexas ou significativas que afetam:
- o desempenho e eficácia originais,
- a segurança ou o uso pretendido do SaMD.
Essas mudanças podem incluir algoritmos novos ou modificados, estruturas de banco de dados, operações.
A seguinte alteração de um SaMD exigiria um novo UDI-PI (não um novo UDI-DI):
- Revisões menores do SaMD serão identificadas com um novo UDI-PI;
- Revisões menores do SaMD são geralmente associadas a correções de bugs, melhorias de usabilidade (não para fins de segurança), patches de segurança ou eficiência operacional.
- Revisões menores devem ser identificadas por métodos de identificação específicos do fabricante (por exemplo, versão, número de revisão, número de série, etc.).
Você pode ver um exemplo de atribuição de UDI ao SaMD na imagem abaixo.

Imagem retirada de IMDRF/UDI WG/N48 FINAL: 2019 - IMDRF – Unique Device Identification system (UDI system)
Quais são as obrigações dos operadores econômicos em relação à UDI?
Fabricantes: serão responsáveis pela atribuição da UDI e pela colocação do portador da UDI, pela apresentação inicial e pelas atualizações das bases de dados da UDI. Os fabricantes devem atualizar o registro relevante do banco de dados no prazo de 30 dias após a alteração de um elemento (prazo para Brasil, UE e EUA), que não exige um novo UDI-DI.
Distribuidores e importadores: devem verificar se, quando aplicável, uma UDI foi atribuída pelo fabricante.
Operadores económicos e instituições de saúde: as autoridades reguladoras podem exigir que os prestadores de cuidados de saúde e as farmácias retalhistas garantam que todos os registos de dispositivos que mantêm incluem a UDI como um componente essencial para permitir a rastreabilidade dos dispositivos. Este período normalmente é equivalente ao período de manutenção dos registros dos pacientes.
Como posso obter um código UDI?
Um UDI pode ser obtido através de uma das agências emissoras oficiais que operam em todo o mundo. As autoridades de saúde credenciam cada agência emissora de UDI naquela região.
|
Base de dados UDI |
||
|
Paises |
Base de dados |
Entidades emissoras aceitas |
|
Brasil |
A definir |
GS1, HIBCC, ICCBBA |
|
União Europeia |
EUDAMED |
GS1, HIBCC, ICCBBA, IFA |
|
Estados Unidos |
GUDID |
GS1, HIBCC, ICCBBA |
Requisito UDI do Brasil
Em 10 de janeiro de 2022 foi publicada a RDC 591/2021. Dispõe sobre a identificação de dispositivos médicos regularizados na ANVISA, por meio do sistema de Identificação Única de Dispositivos Médicos (UDI).
Em 2 de julho de 2024 foi publicada a RDC 884/2024, de 26 de junho de 2024, que altera a RDC 591/2021 e prorroga os prazos para atribuição da UDI aos dispositivos médicos das classes II, III e IV por mais um ano. O prazo para os dispositivos médicos classe I permanece inalterado.
|
Cronograma de conformidade no Brasil |
|
|
Classe de risco do dispositivo |
Datas de conformidade (rotulagem) |
|
Classe IV |
10 julho 2025 |
|
Classe III |
10 janeiro de 2026 |
|
Classe II |
10 Janeiro de 2027 |
|
Classe I |
10 Janeiro de 2028 |
Requisitos UDI da União Europeia
Para melhorar a transparência e o acesso à informação, foi desenvolvida a base de dados europeia sobre dispositivos médicos (EUDAMED). Inicialmente, os fabricantes não eram obrigados a registar os seus dispositivos médicos na EUDAMED até que todos os seus seis módulos tivessem sido configurados. No entanto, embora já estejam disponíveis três módulos e se espere que mais dois estejam disponíveis em 2024, é pouco provável que o módulo final esteja concluído até final de 2027.
Embora a Comissão Europeia não esteja em posição de exigir a utilização do módulo de registo UDI/Dispositivos até que o EUDAMED esteja totalmente funcional, os fabricantes já podem introduzir informações UDI/Dispositivo no sistema numa base voluntária.
|
Cronograma de conformidade da União Europeia |
|
|
Classe de risco do dispositivo |
Datas de conformidade (rotulagem) |
|
Dispositivos implantáveis e classe III |
26 de maio de 2023 |
|
dispositivos implantáveis de classe III e classe IIb, ‘dispositivos antigos’ |
31 de dezembro de 2027 |
|
Classe IIa e IIb |
26 de maio de 2023 |
|
Classe I |
26 de maio de 2025 |
|
Implantável e Classe III – reutilizável |
26 de maio de 2023 |
|
Classe IIa e IIb - reutilizáveis |
26 de maio de 2025 |
|
Dispositivos de classe IIb e classe IIa, classe Im e classe Is ‘dispositivos legados’ |
31 de dezembro de 2028 |
|
Classe I - reutilizável |
26 de maio de 2027 |
|
Classe D (DIV) |
26 de maio de 2023 |
|
Classe B e C (IVD) |
26 de maio de 2025 |
|
Classe A (DIV) |
26 de maio de 2027 |
Requisitos UDI dos Estados Unidos
O FDA exige que todas as informações da UDI sejam inseridas no banco de dados GUDID específico dos EUA.
|
Cronograma de conformidade dos Estados Unidos |
|
|
Classe de risco do dispositivo |
Datas de conformidade (rotulagem) |
|
Classe III |
24 de setembro de 2014 |
|
Classe II |
24 de setembro de 2016 |
|
Classe II - reutilizável |
24 de setembro de 2018 |
|
Classe I |
24 de setembro de 2018 |
|
Classe I - reutilizável |
9 de dezembro de 2022 |
Implementação do projeto UDI – principais desafios para profissionais de validação
As empresas fabricantes e importadoras deverão planejar a aquisição, qualificação, validação e tecnologia das soluções que farão parte do projeto UDI.
Como você deve saber, as empresas de dispositivos médicos são altamente regulamentadas e precisam provar que seu desenvolvimento, processos e produto final são consistentes. Este é o papel da validação e qualificação.
A realização da validação minimizará os riscos do projeto e garantirá a conformidade da aplicação, evitando sanções dos órgãos reguladores.
Quanto mais cedo as empresas se prepararem, mais fácil será a disponibilidade e o planeamento.
Embora possa parecer que há tempo suficiente para se preparar (uma vez que a EUDAMED não está totalmente funcional), é importante reconhecer que existem inúmeras tarefas complexas que devem ser concluídas antes do prazo.
Para países como o Brasil, embora a base de dados ainda não tenha sido definida, todos os demais requisitos devem ser atendidos, ou seja, as empresas devem demonstrar conformidade com seus processos, sua tecnologia e seu cenário de informação.
O software/equipamento utilizado na implementação do sistema UDI (por exemplo, sistema utilizado para rotulagem UDI, sistema de visão, sistema para carregamento automático de dados na base de dados UDI) deve permanecer validado de acordo com os regulamentos relevantes.
Um dos maiores desafios para as pequenas e médias indústrias é a escassez de profissionais altamente qualificados na área de validação.
Muitas vezes, a formação de profissionais nessa área leva tempo, o que pode atrasar o registro e a produção do produto.
O projeto UDI envolve muita tecnologia; porém, muitas empresas ainda optam por realizar a validação da forma tradicional, no papel, o que torna o processo demorado e burocrático.
Este projeto possui características diferentes, pois possui diversas etapas, de acordo com a classe de risco do dispositivo, isso significa que suas respectivas validações poderão ocorrer em momentos distintos do projeto.
A validação sem papel é a melhor solução e estratégia. A validação não deve ser o “gargalo” da cadeia de inovação e tecnologia, mas sim uma aliada para garantir a conformidade em cada etapa da implementação e aplicação.
Resumidamente, cada classe de risco de dispositivo que será inserida poderá eventualmente incluir novos equipamentos e sistemas e ter regras ou requisitos de negócio diferentes.
Esse comportamento é típico de um projeto que requer liberações em fases e vários relatórios de validação em momentos diferentes.
GO!FIVE® possui uma função de liberação parcial para facilitar projetos que possuem um único Plano de Validação e uma única estratégia, mas fases de liberação distintas.
Algumas vantagens
Como forma de viabilizar o projeto, contamos com soluções completas com especialistas (prestação de serviços) e software (solução de validação paperless) para apoiar projetos em qualquer lugar do mundo.
GO!FIVE® é uma plataforma SaaS escalável, onde é possível validar 7x mais rápido seguindo o método ágil. Possui mais de 17 anos de conhecimento de consultoria em software com validações pré-formatadas integradas (risco, requisitos e testes).
GO!FIVE® atende aos requisitos da FDA, EMA e OMS e permite projetos ágeis:
- Importar modelos de validação para projeto UDI (requisitos, riscos e testes)
- Validação 7x mais rápida
- Capacidade de IA para organizar documentos PDF
- Criar/definir itens por matriz linear
- Liberações parciais
- Facilidade de manutenção de status validado
- Testar a funcionalidade de replicação
- A única solução de validação com conteúdo qualitativo no banco de dados





{kind=link}
{kind=link}
{kind=link}