
¿Qué es UDI (Identificación única de dispositivo)? Principios para el diseño y operación de sistemas – dispositivos médicos

¿Qué es UDI (Identificación única de dispositivo)?
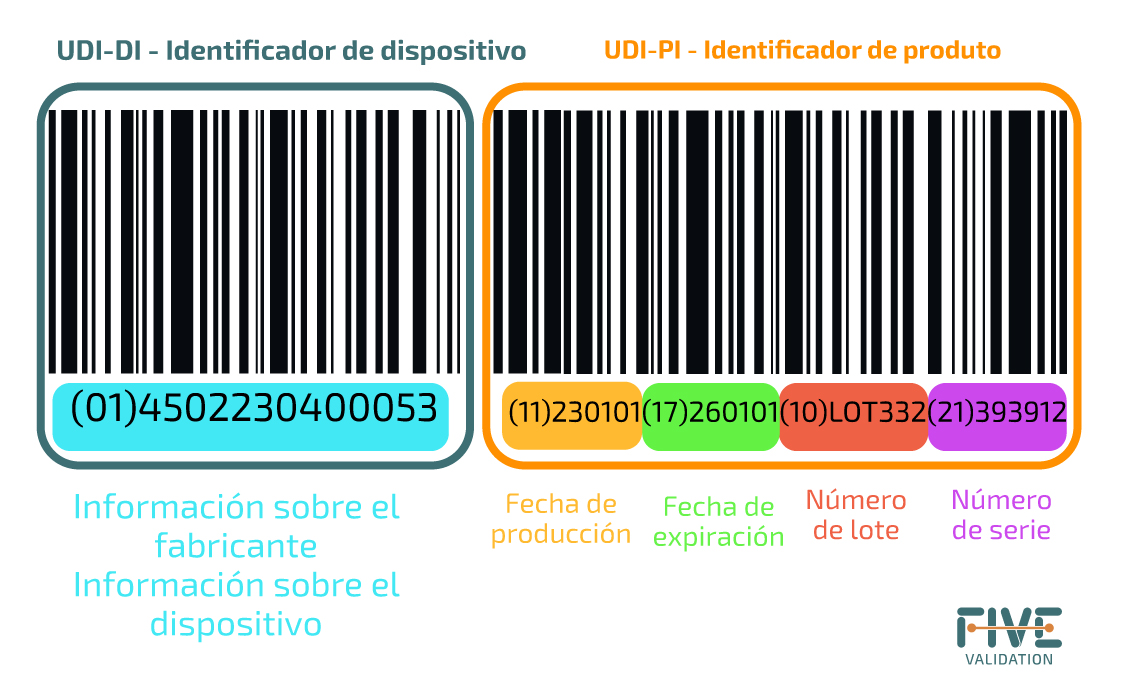
Nota: PI (Identificador de Producción) es un código variable que representa uno o más de los siguientes elementos: número de serie del dispositivo; número de lote; fecha de vencimiento del dispositivo; fecha de fabricación del dispositivo presentada en formato estándar (AAAA-MM-DD); Código de identificación en el caso de un producto a base de células humanas (HCT/P).
La siguiente figura describe los pasos de la implementación de la UDI.

Los estándares y plazos de la UDI varían según el mercado y el producto, y han sido revisados varias veces en algunos países.
Aunque en Europa las directrices comenzaron con la publicación del IMDRF en 2013, de la que hablaremos más adelante, esto no fue nada nuevo para la industria de dispositivos médicos, ya que ha sido una necesidad para los productos exportados a EE. UU. durante muchos años.
En Europa, el número UDI también será un requisito para el seguimiento del producto, para lo cual se deberá utilizar la base de datos EUDAMED.
A diferencia de EE. UU. y Brasil, para Europa se requiere un Basic-UDI-DI. Basic-UDI-DI (identificación única básica de dispositivo - Identificador de dispositivo) es un identificador de agrupación de dispositivos médicos con el mismo uso previsto, clase de riesgo y características esenciales de diseño y fabricación. A diferencia de UDI-DI, que es específico para cada modelo y versión de dispositivo, Basic-UDI-DI no aparece en la etiqueta ni en el embalaje del producto. En cambio, sirve como identificador clave en bases de datos y documentación regulatorias, asegurando la trazabilidad y facilitando los procesos.
Importante: La empresa regulada debe asegurarse de que los sistemas UDI cumplan con la normativa adecuada. Ésta es la función del proceso de validación. ¡Explore nuestra página de servicios y descubra cómo FIVE Validation puede mejorar su trabajo!
International Medical Device Regulators Forum (IMDRF)
¿Qué hay en la etiqueta?
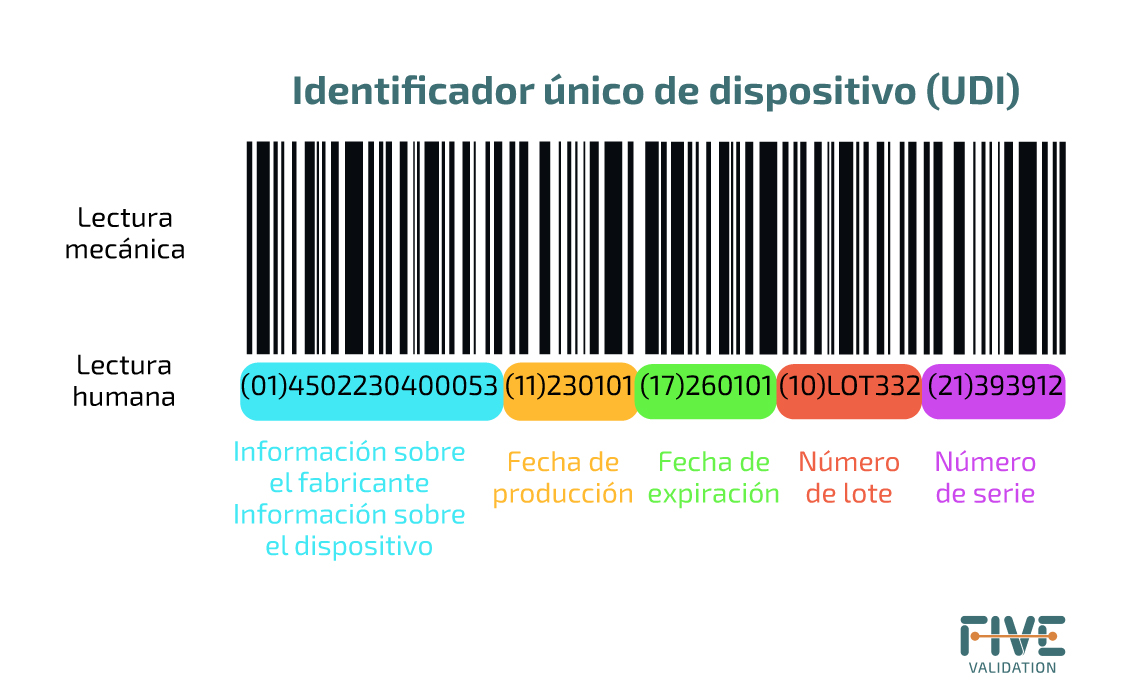
Algunos de los números entre paréntesis en el componente UDI-PI serán reconocibles. Cada uno de estos números transmite detalles específicos sobre el artículo:
(01) GTIN
(10) es para el número de lote.
(11) es para la fecha de producción.
(17) define la fecha de vencimiento.
(21) es el número de serie.
Esto significa que con cada nuevo lote o fecha de vencimiento actualizada, todos estos detalles cambiarán, haciendo que el código sea dinámico.
La UDI se compone de dos partes:
Identificador de Dispositivo (UDI-DI)
+
Identificador de Producción (UDI-PI)
a) El número de lote dentro del cual se fabricó un dispositivo;
b) El número de serie de un dispositivo específico;
c) La fecha de vencimiento de un dispositivo específico;
d) La fecha de fabricación (puede no ser requerida si existen otros Identificadores de Producción en la etiqueta);
e) La versión, para Software como Dispositivo Médico (SaMD);
Nota: La versión SaMD puede capturarse en el identificador de producción del lote según ciertas regulaciones nacionales.
f) El Código de Identificación Distinto (DIC), cuando corresponda. Este número es un identificador esencial para productos médicos de origen humano.
Nota: Los productos médicos de origen humano (MPHO) incluyen sangre, órganos, médula ósea, sangre del cordón umbilical, córneas, tejidos, células reproductivas y leche derivada de humanos para uso terapéutico. El uso de DIC en UDI se limita a productos MPHO regulados como dispositivos médicos.
a) Identificación automática y captura de datos (AIDC): es una tecnología utilizada para la captura automática de datos. Las tecnologías AIDC incluyen códigos de barras, tarjetas inteligentes, biometría y RFID (identificación por radiofrecuencia).
b) Interpretación legible por humanos (HRI): es una interpretación legible por humanos de los caracteres de datos codificados en el soporte UDI. Es importante si no existe un sistema de lectura automática de códigos.
Por lo tanto, el código UDI debe tener inicialmente una sección legible por sí mismo, como un código de barras, y estar seguido de una parte legible por humanos, como códigos numéricos.
Cuando el sistema UDI esté completamente implementado, la etiqueta de la mayoría de los dispositivos incluirá UDI legible por humanos y soporte AIDC.
El sistema UDI pretende ser un identificador que también se utiliza en el contexto de transacciones comerciales y clínicas, incluida la trazabilidad de los dispositivos en el entorno posterior a la comercialización (por ejemplo, órdenes de compra, facturas, gestión de mantenimiento/inventario, notas clínicas, etc.).
El soporte UDI deberá colocarse en la etiqueta del dispositivo y en todos los niveles superiores del embalaje y, en el caso de dispositivos reutilizables, en el propio dispositivo (marcado directo).
¿Le gustaría saber más sobre la función de validación en el proyecto UDI? Póngase en contacto con uno de nuestros expertos en [email protected]
¿Cuáles son las obligaciones de los operadores económicos respecto de la UDI?
La UDI debe asignarse al nivel del sistema Software como dispositivo médico (SaMD).
El número de versión de SaMD se considera el mecanismo de control de fabricación y debe mostrarse en el UDI-PI.
El siguiente cambio a un SaMD requeriría un nuevo UDI-DI:
- Las revisiones importantes de SaMD se identificarán con un nuevo UDI-DI;
- Las principales revisiones del SaMD se entienden como cambios complejos o significativos que afectan a:
- el rendimiento y la eficacia originales,
- la seguridad o el uso previsto de SaMD.
Estos cambios pueden incluir algoritmos, estructuras de bases de datos y operaciones nuevos o modificados.
El siguiente cambio a un SaMD requeriría un nuevo UDI-PI (no un nuevo UDI-DI):
- Las revisiones menores de SaMD se identificarán con un nuevo UDI-PI;
- Las revisiones menores de SaMD generalmente están asociadas con correcciones de errores, mejoras de usabilidad (no con fines de seguridad), parches de seguridad o eficiencia operativa.
- Las revisiones menores deben identificarse mediante métodos de identificación específicos del fabricante (por ejmplo, versión, número de revisión, número de serie, etc.).
Puede ver un ejemplo de asignación de UDI a SaMD en la imagen a continuación.

Imagen copiada de IMDRF/UDI WG/N48 FINAL: 2019 - IMDRF – Unique Device Identification system (UDI system)
¿Cuáles son las obligaciones de los operadores económicos respecto de la UDI?
Fabricantes: serán responsables de la asignación de la UDI y la colocación del titular de la UDI, el envío inicial y las actualizaciones de la base de datos de la UDI. Los fabricantes deben actualizar el registro de la base de datos correspondiente dentro de los 30 días posteriores al cambio de un elemento (plazo para Brasil, UE y EE. UU.), lo que no requiere una nueva UDI-DI.
Distribuidores e importadores: deberán verificar que, en su caso, se haya asignado una UDI por parte del fabricante.
Operadores económicos e instituciones de atención médica: las autoridades reguladoras pueden exigir a los proveedores de atención médica y a las farmacias minoristas que se aseguren de que todos los registros de dispositivos que mantienen incluyan la UDI como un componente esencial para permitir la trazabilidad de los dispositivos. Este período normalmente equivale al período de mantenimiento de registros de pacientes.
¿Cómo puedo obtener un código UDI?
Se puede obtener una UDI a través de una de las agencias emisoras oficiales que operan en todo el mundo. Las autoridades sanitarias acreditan a cada organismo emisor de la UDI en esa región.
|
Base de datos UDI |
||
|
Países |
Base de datos |
Entidades emisoras aceptadas |
|
Brasil |
A definir |
GS1, HIBCC, ICCBBA |
|
Unión Europea |
EUDAMED |
GS1, HIBCC, ICCBBA, IFA |
|
Estados Unidos |
GUDID |
GS1, HIBCC, ICCBBA |
Requisito de UDI de Brasil
El 10 de enero de 2022 se publicó la RDC 591/2021. Prevé la identificación de dispositivos médicos regulados por la ANVISA, a través del sistema de Identificación Única de Dispositivos Médicos (UDI).
El 2 de julio de 2024 se publicó la RDC 884/2024, de 26 de junio de 2024, que modifica la RDC 591/2021 y amplía por un año más los plazos para la atribución de la UDI a los productos sanitarios de las clases II, III y IV. El plazo para los productos sanitarios de clase I se mantiene sin cambios.
|
Cronograma de cumplimiento en Brasil |
|
|
Clase de riesgo del dispositivo |
Fechas de cumplimiento (etiquetado) |
|
Clase IV |
10 de julio de 2025 |
|
Clase III |
10 de enero de 2026 |
|
Clase II |
10 de enero de 2027 |
|
Clase I |
10 de enero de 2028 |
Requisitos UDI de la Unión Europea
Para mejorar la transparencia y el acceso a la información, se desarrolló la base de datos europea sobre productos sanitarios (EUDAMED). Inicialmente, los fabricantes no estaban obligados a registrar sus dispositivos médicos en EUDAMED hasta que se hubieran configurado sus seis módulos. Sin embargo, aunque ya hay tres módulos disponibles y se espera que dos más estén disponibles en 2024, es poco probable que el módulo final esté terminado para finales de 2027.
Aunque la Comisión Europea no está en condiciones de exigir el uso del módulo de registro de dispositivo/UDI hasta que EUDAMED sea completamente funcional, los fabricantes ahora pueden ingresar información de dispositivo/UDI en el sistema de forma voluntaria.
|
Cronograma de cumplimiento de la Unión Europea |
|
|
Clase de riesgo del dispositivo |
Datas de conformidade (rotulagem) |
|
Dispositivos Implantables y Clase III |
26 de maio de 2023 |
|
Dispositivos implantables clase III y clase IIb, “dispositivos antiguos” |
31 de dezembro de 2027 |
|
Clase IIa y IIb |
26 de maio de 2023 |
|
Clase I |
26 de maio de 2025 |
|
Implantable y Clase III – reutilizable |
26 de maio de 2023 |
|
Clase IIa y IIb - reutilizable |
26 de maio de 2025 |
|
Dispositivos Clase IIb y Clase IIa, Clase Im y Clase Is 'dispositivos antiguos' |
31 de dezembro de 2028 |
|
Clase I - reutilizable |
26 de maio de 2027 |
|
Clase D (DIV) |
26 de maio de 2023 |
|
Clase B y C (IVD) |
26 de maio de 2025 |
|
Clase A (DIV) |
26 de maio de 2027 |
Requisitos UDI de Estados Unidos
La FDA exige que toda la información UDI se ingrese en la base de datos GUDID específica de EE. UU.
|
Cronograma de cumplimiento de Estados Unidos |
|
|
Clase de riesgo del dispositivo |
Fechas de cumplimiento (etiquetado) |
|
Clase III |
24 de septiembre de 2014 |
|
Clase II |
24 de septiembre de 2016 |
|
Clase II - reutilizable |
24 de septiembre de 2018 |
|
Clase I |
24 de septiembre de 2018 |
|
Clase I - reutilizable |
9 de diciembre de 2021 |
Implementación del proyecto UDI – principales desafíos para los profesionales de la validación
Las empresas fabricantes e importadoras deberán planificar la adquisición, calificación, validación y tecnología de las soluciones que formarán parte del proyecto UDI.
Como sabrá, las empresas de dispositivos médicos están altamente reguladas y necesitan demostrar que su desarrollo, procesos y producto final son consistentes. Éste es el papel de la validación y la calificación.
La realización de la validación minimizará los riesgos del proyecto y garantizará el cumplimiento de la aplicación, evitando sanciones por parte de los organismos reguladores.
Cuanto antes se preparen las empresas, más fácil será la disponibilidad y la planificación.
Si bien puede parecer que hay tiempo suficiente para prepararse (ya que EUDAMED no es completamente funcional), es importante reconocer que existen numerosas tareas complejas que deben completarse antes de la fecha límite.
Para países como Brasil, aunque la base de datos aún no esté definida, todos los demás requisitos deben cumplirse, es decir, las empresas deben demostrar el cumplimiento de sus procesos, su tecnología y su escenario de información.
El software/equipo utilizado en la implementación del sistema UDI (por ejemplo, sistema utilizado para el etiquetado UDI, sistema de visión, sistema para la carga automática de datos en la base de datos UDI) debe permanecer validado de acuerdo con las regulaciones pertinentes.
Uno de los principales desafíos para las pequeñas y medianas industrias es la falta de una fuerza laboral de validación altamente calificada.
La formación de profesionales en esta área suele llevar tiempo, lo que puede retrasar el registro y la producción del producto.
El proyecto UDI involucra mucha tecnología; sin embargo, muchas empresas todavía optan por realizar la validación de forma tradicional, en papel, lo que hace que el proceso sea lento y burocrático.
Este proyecto tiene diferentes características, ya que tiene varias etapas, dependiendo de la clase de riesgo del dispositivo, lo que significa que sus respectivas validaciones pueden ocurrir en diferentes momentos del proyecto.
La validación sin papel es la mejor solución y estrategia. La validación no debe ser el “cuello de botella” en la cadena de innovación y tecnología, sino más bien un aliado para asegurar el cumplimiento en cada etapa de implementación y aplicación.
En resumen, cada clase de riesgo de dispositivo que se insertará puede eventualmente incluir nuevos equipos y sistemas y tener reglas o requisitos comerciales diferentes.
Este comportamiento es típico de un proyecto que requiere lanzamientos por fases y múltiples informes de validación en diferentes momentos.
GO!FIVE® tiene una función de lanzamiento parcial para facilitar proyectos que tienen un único Plan de Validación y una única estrategia, pero distintas fases de lanzamiento.
Algunas ventajas
Como forma de viabilizar el proyecto, contamos con soluciones completas con expertos (prestación de servicios) y software (solución de validación sin papel) para soportar proyectos en cualquier parte del mundo.
GO!FIVE® es una plataforma SaaS escalable, donde es posible validar 7 veces más rápido siguiendo el método ágil. Tiene más de 17 años de conocimiento en consultoría de software con validaciones integradas preformateadas (riesgo, requisitos y pruebas).
GO!FIVE® cumple con los requisitos de la FDA, la EMA y la OMS y permite proyectos ágiles:
- Importar modelos de validación para proyecto UDI (requisitos, riesgos y pruebas)
- Validación 7 veces más rápida
- Capacidad de IA para organizar documentos PDF
- Crear/definir elementos por matriz lineal
- Lanzamientos parciales
- Facilidad para mantener el estado validado
- Probar la funcionalidad de replicación
- La única solución de validación con contenido cualitativo en la base de datos.
Sobre el autor: Lílian Ribeiro

Es ingeniería química con 10 años de experiencia técnica y comercial en la industria alimentaria, especializándose en calidad corporativa/control de calidad. Además, tiene experiencia en el sector sanitario/farmacéutico.
Como entusiasta de la validación sin papel, le apasiona aportar eficiencia e innovación a las empresas de ciencias biológicas.
Su experiencia radica en proyectos de validación y calificación, incluidos VLMS, ERP, EQMS, automatización (PW) y calificación de infraestructura de TI.
Sobre el revisor: Sílvia Martins

Ingeniera eléctrica, con +20 años de experiencia al servicio de empresas (bio)farmacéuticas y de dispositivos médicos. Graduada en Inglaterra en GAMP5® y FDA 21 CFR Part11, en Alemania en validación SAP® y Dinamarca en Data Integrity and Governance. Como CEO y cofundadora de FIVE Validation, una empresa comprometida con la racionalización de los procesos de cumplimiento, Silvia se dedica a agilizar los procedimientos para los clientes, manteniendo un alto nivel de solidez y cumplimiento.



{kind=link}
{kind=link}
{kind=link}